




元々は企業が、原薬や中間体、製剤原料、医薬品添加剤などの情報を米食品医薬品局(FDA)に任意で登録する米国の制度を指す。中国では同様の制度があり、医薬品包装・医薬品添加物と2019-56号公告によって定められている。
医薬品包装・医薬品添加物と2019-56号公告
- < 医薬品包装(中国語:薬包材)とは >
-
医薬品の生産または処方の際に、医薬品と直接接触する包装材料、または容器を示す。
- < 医薬品添加物(中国語:薬用補料)とは >
-
医薬品の生産または処方の際に使われる賦形剤・添加剤などを示し、医薬品の活性成分を除き、安全性において合理的に評価され、且医薬品製剤の成分に含まれる物質。
- 国家医薬品管理局によるさらなる医薬品関連事項改善のための審査承認およびその管理作業に関する事項の報告書(2019年第56号)一.全体要求(一)より抜粋
旧国家食品薬品監督管理総局(CFDA)は、中国共産党中央弁公庁と国務院弁公庁の「医薬品医療機器イノベーションの推奨及び審査承認制度改革の深化に関する意見」を実施するために、「原薬、医薬品添加物(以下、「添加物」という)、医薬品包装材料(以下、「包装材料」という)の審査承認事項の調整に関する公告」(2017年第146号)を発表した。このたび、原薬、添加物、医薬品と直接接触する包装材料及び容器(以下、「原薬等」という)並びに医薬品製剤(以下、「製剤」という)の関連審査承認及び監督管理関連事項をさらに明確にするために、以下に公告する。
医薬品包装・医薬品添加物の登記制度について
医薬品承認審査を簡素化し、医薬品再登記制度を整備させる目的とし、法令の改定を重ね、現時点では下記文書が最新の改定。

- 医薬品包装・医薬品添加物の概念を明確化し、既存の医薬品製剤や医療器から切り離し、単独の登記制度にて運用する。
- 医薬品と医薬品包装、医薬品添加物との関連審査を行い、医薬品包装と医薬品添加物に対する独自的な承認審査を医薬品登記申請の時に一緒に承認審査を行う。
上記2017-42号の実務規定として2019年8月に施行されたのが2019年56号
新旧制度の違いについて

従来の制度:
包装材や添加物に対して独自な承認審査をしていました。
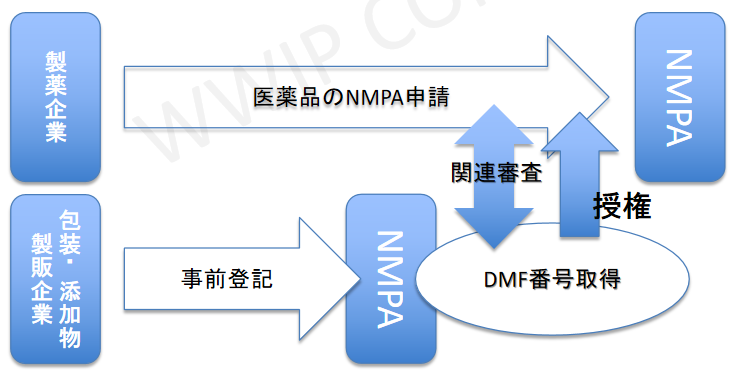
新制度:
包装材と添加物は登記性(届出)になり、製品情報をデーターベースに登記し、DMF番号を取得。
製薬企業が医薬品NMPA申請する際に該当DMF番号と関連審査を行います。
56号登記:制度対象の製品
薬包材
(医薬品包装材料)
- 中国語:薬包材
- 日本語:医薬品包装材料
医薬品を入れるためのもので、医薬品と直接接触するもの。例えば錠剤や注射剤などの容器・栓・蓋、乾燥剤などが該当する。
薬用補料
(医薬品添加物)

- 中国語:薬用補料
- 日本語:医薬品添加物
製剤に含まれる有効成分以外の物質、賦形剤、安定剤、保存剤、緩衝剤、矯味剤、懸濁化剤、乳化剤、溶解補助剤、着色剤など。
原料薬(原薬)
- 中国語:原料薬
- 日本語:原薬
医薬品の有効成分に該当する低分子化合物など。例えば抗生剤の中には、細菌を殺傷・増殖阻害するための有効成分が入っています。
56号登記:全体的な要件
【一】原薬等の使用は、医薬品関係様式事項に適合しなければならない。
それは主に、原薬等の品質、安全性及び効能が、製剤のニーズを満たさなければならないことを指す。原薬等と製剤の関連審査承認は、原薬等登記者が登記プラットフォームに登記し、製剤登記申請者が登記申請を行う際にプラットフォームの登記情報と関連づけることになる。
特別な理由のために原薬等をプラットフォームに登記することはできない場合には、製剤の登記申請時に、製剤登記申請者が原薬等の研究データを提供することもできる。
- 原薬等の事前登記を要求
- 製剤の申請者は事前登記された登記番号とともに製剤を申請
- 但し、例外規定あり
【二】原薬等登記者は、責任をもって登記プラットフォームにおける登記情報の更新を行い、登記情報の信憑性及び完全性について責任を負わなければならない。
国内の原薬等の供給者は、原薬等の登記者として自ら製品登記しなければならない。
国外の原薬等の供給者は、中国駐在代表者又は中国代理申請会社に委託して登記することができ、登記資料は中国語でなければならず、国外原薬等の供給者及び中国代理会社は、登記資料の信憑性と完全性に共同で責任を負わなければならない。
- 海外からは直接申請することはできない
- 原薬等の生産企業の中国支社または中国国内の代理申請会社を通じて行う必要がある
- 中国国内の代理申請会社は、海外企業と共に申請資料に責任を持つ
(十八)国外の原薬等供給者が登録代理人を変更する場合は、関連書類を提出することで変更することができる。
資料には、現行の法令に基づき変更理由の説明書、国外の原薬等供給者からの委任状(公証文書とその中国語訳)、新しい代理人の営業許可証のコピー、国外の原薬等供給者が元代理人との委託関係を解除する文書(公証文書との中国語訳)を含む。
【三】製剤登記申請者は、製剤登記申請を行う際に、原薬等登記番号及び原薬等登記者の使用授権書を提出しなければならない。
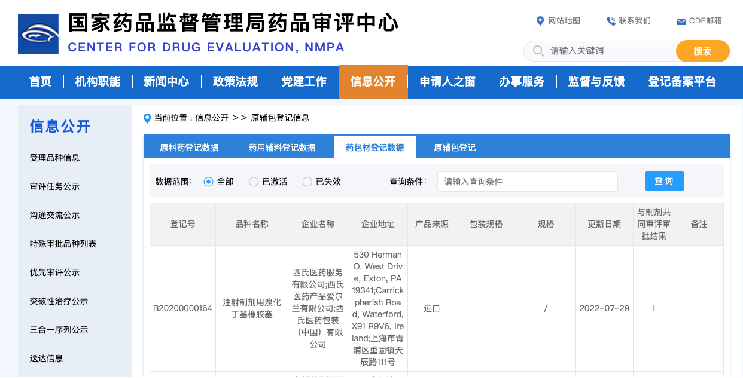
- 登記が完了するとデータベースに公開され、登記番号が振られる
- 製剤申請の際には、授権を受けて登記番号を用いて申請
- データベースをみた中国企業からの問い合わせがあることも
【七】 製剤登録申請と登録した原薬等は関連を持つ。
製剤が承認されると、関連する原薬等も技術審査を通過し、登録プラットフォーム上で「A」と表示される。
技術審査に合格していないものや、製剤登録されていないものには、「I」と表示される。
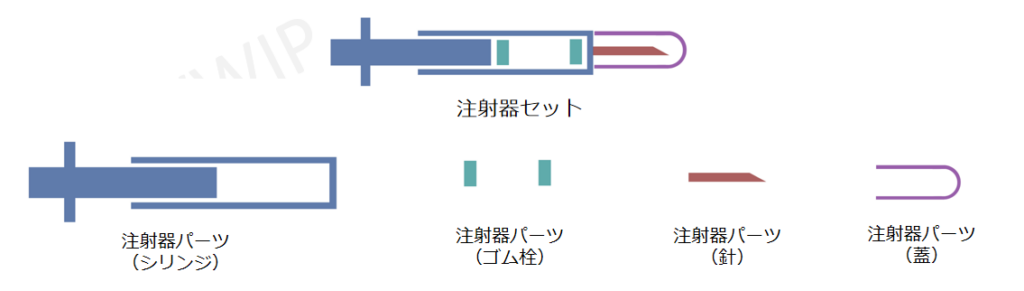
(十一)添加物、包装材料は行政許可を廃止とし、無料でプラ
56号登記:申請要求
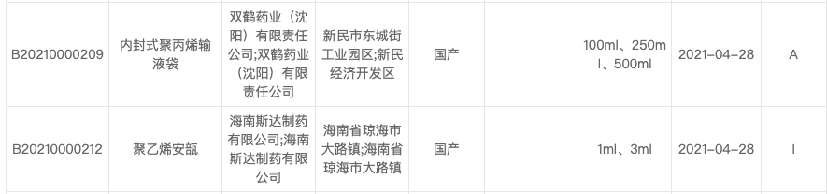
薬包材における「セット登記」と「パーツ登記」
医薬品包装はセット(組合件)とパーツ(組件)2種の申請パターン。
セットは各パーツによって構成される。
- セット
-
YBB00112004: 事前装填型注射器
- パーツ
-
YBB00062004: シリンジ部分、YBB00092004: 針部分、YBB00102004: 蓋部分
-
YBB00072004:クロロブチルゴム部分、YBB00082004: ブロモブチルゴム部分
添付① 医薬品添加物登録データの要件
- 添加物の分類
- 国内外で販売されている薬品中に使用された履歴のないもので下記に該当するもの
- 1.1新しい分子構造の添加物及び第1.2、1.3に含まれない添加物;
- 1.2既に使用の履歴がある添加物に簡単な化学構造の変化を加えたもの(例:塩基、水和物等);
- 1.3二者及び二者以上の既に使用の履歴のある添加物をコプロセッシングして得られた添加物;
- 1.4既に使用の履歴があるが投与経路に変化が生じた添加物。
- 国内外で販売されている薬品中に使用された履歴のあるもので且つ下記に該当するもの
- 2.1中国薬局方/USP/EP/BP/JPの何れにも収載のない添加物;
- 2.2USP/EP/BP/JPの内一つに収載があるが、国内で販売されている薬品中に使用されたことがない添加物;
- 2.3USP/EP/BP/JPの内一つに収載があるが、中国薬局方に未収載の添加物;
- 2.4中国薬局方に既に収載されている添加物。
- 食品或いは化粧品中に既に使用された履歴のあるもので且つ下記に該当するもの
- 3.1食品安全国家標準に合致したもので経口剤に用いられる添加物;
- 3.2化粧品国家標準或いは業界標準に合致したもので外用薬に用いられる添加物。
- 国内外で販売されている薬品中に使用された履歴のないもので下記に該当するもの
- 申請必要資料
- 登記者の基本情報
- 登記者名称、住所、生産拠点
- 証明書類
- 研究資料の保管場所住所
- 添加物の基本情報
- 名称
- 構造と組成
- 物理的・化学的特性及び基本特性
- 国内外における販売及び使用承認に関する情報
- 国内外の薬局方への収載状況
- 生産情報
- 生産工程及び工程管理
- 原料の管理
- 重要工程及び中間体の管理
- 工程の検証と評価
- 生産工程の開発
- 特性の鑑定
- 構造と物理的・化学的性質に関する研究
- 不純物に関する研究
- 機能特性
- 品質管理
- 品質規格
- 分析方法の検証
- 品質規格設定における根拠
- ロット検査の報告
- 安定性に関する研究
- 安定性に関する要約
- 安定性を示すデータ
- 添加物の包装
- 薬理学・毒理学的研究
- 登記者の基本情報
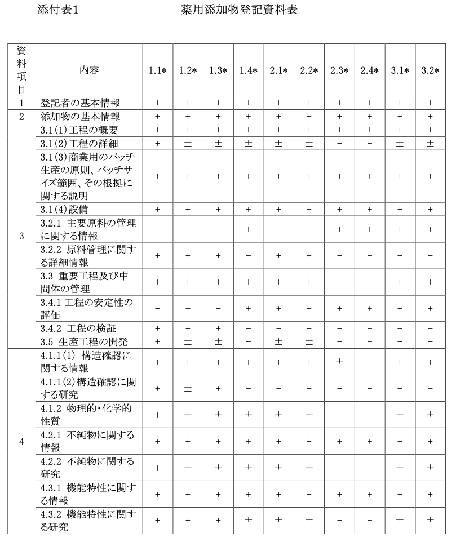
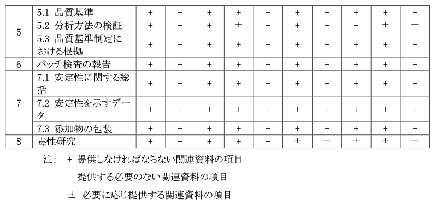
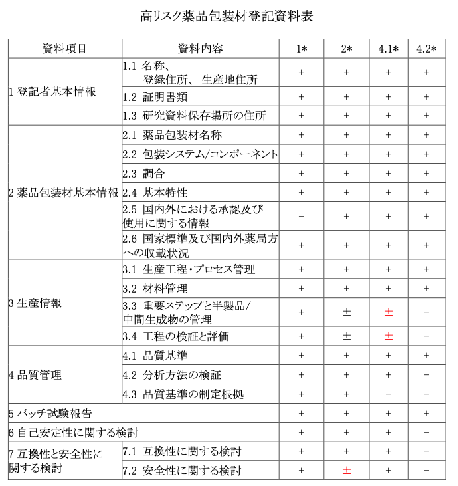
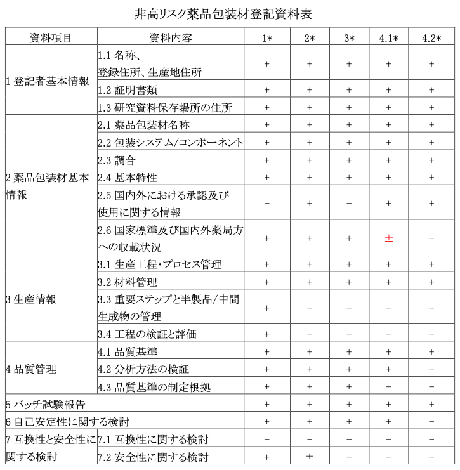
添付② 薬品包装材の登記資料に関する要件
- 使用状況による分類:
- 国内外で販売されている薬品において使用されたことのない薬品包装材(新材料・新構造等);
- 国内外で販売されている薬品において既に使用されたことがあるが、薬品の投与経路に変更があり且つリスクが上昇する薬品包装材;
- 国内外で販売されている薬品において使用されたことがないが、食品包装中に食品と直接接触する形で使用されたことが証明できる薬品包装材(経口剤に限る);
- 販売されている薬品で同一の投与経路で既に使用されたことのある薬品包装材
- 登録証の無い薬品包装材
- 登録証の有る薬品包装材
- その他
- 申請必要資料
- 登記者基本情報
- 名称、住所、製造工場、生産地住所
- 証明書類
- 研究資料保存場所の住所
- 薬品包装材基本情報
- 薬品包装材の名称
- 包装システム/コンポーネント
- 調合
- 基本特性
- 国内外における販売及び使用の承認に関する情報
- 国家標準及び国内外薬局方への収載状況
- 生産情報
- 生産工程・プロセス管理
- 材料管理
- 重要ステップと半製品/中間生成物の管理工程の検証と評価
- 品質管理
- 品質基準
- 分析方法の検証
- 品質規格設定にあたっての根拠
- ロット試験報告
- 自己安定性に関する検討
- 互換性と安全性に関する検討
- 互換性に関する検討
- 安全性に関する検討
- 登記者基本情報
添付③ 登記が免除される製品リスト
薬品製剤時で使用される一部のフレーバー・香料・色素・pH調整剤等の薬品添加物は146号公告の要件に従わず、登記をおこなうことができる。具体的には下記の通り:
- フレーバー(甘味剤):例としてスクロース、単シロップ、マンニトール、ソルビトール、サッカリンナトリウム、アスパルタン、スクラロース、ステビオシド、グルコース、キシリトール、マルチトール等がある。当該添加物は製剤中に矯味剤(甘味剤)として使用される場合に限る。
- 香料:例として、オレンジエッセンス、バナナエッセンス、バニリン等がある。食品標準を満たすためには、現行版のGB2760《食品安全国家標準食品添加剤使用標準》、GB30616《食品安全国家標準食品用香精》及びGB29938《食品安全国家標準食品用香料通則》等、およびその他の関連要件に準拠する必要がある。
- 色素(着色剤):例として酸化鉄、植物性カーボンブラック、コチニールレッド等がある。食品標準を満たすためには、現行版のGB2760《食品安全国家標準食品添加剤使用標準》等、およびその他の関連要件に準拠する必要がある。
- pH調整剤(注射剤中に用いるpH調節剤を含む):例としてリンゴ酸、フマル酸、酢酸、酢酸ナトリウム、クエン酸(ナトリウム、カリウム塩)、酒石酸、水酸化ナトリウム、強塩基溶液、塩酸、硫酸、リン酸、乳酸、リン酸二水素カリウム、リン酸水素ニカリウム、リン酸水素二ナトリウム、リン酸二水素ナトリウム等がある。
- 補助材としてのみ使用し、調製準備工程が簡潔で、物理的・化学的性質が安定している無機塩類(注射剤中に用いる無機塩類を含む):例として炭酸カルシウム、炭酸ナトリウム、塩化カリウム、塩化カルシウム、塩化マグネシウム、リン酸カルシウム、リン酸水素カルシウム、硫酸カルシウム、炭酸水素ナトリウム)等がある。
- 経口剤の印字に使用するベンゼンフリーインク。
添付④ 医薬品原料・添加物・医薬品包装材料の年次報告に関する基本要件
医薬品原料・添加物、医薬品包装材料の年次報告書に関する基本要件
- 原添包(原料・添加物・包装材料)登録者は毎年の第一四半期に申請者の窓口を介して前年の年次報告書を提出しなければならない。
- 年次報告書中には前年度の製品変更に関する総括を含めなければならず、如何なる変更も無い場合はその旨の声明を含めなければならない。
- 年次報告書中には企業名称、薬品名称等の関連する製剤製品の情報を含めなければならない。
登記の流れ
製品の初歩的な情報を分析の上、見積もりを提出します。双方合意の上、NDA締結し、進めていきます。
提示いただいた資料を中国の規定に従い確認。追加資料の要否等を確認します。
提示いただいた資料を中国語翻訳し、NMPAが規定された形式で申請資料を作成します。
NMPAに申請資料を提出します。
審査が開始されます。
審査が通過し、DMF番号を取得します。
製薬企業がDMF情報を基に関連審査が可能になります。
お気軽にお問い合わせください
お電話でお問い合わせは
03-6206-1723